自身免疫(autoimmunity)是指机体免疫系统对自身抗原发生免疫应答,产生自身抗体和(或)自身致敏淋巴细胞的现象。自身耐受(self tolerance)是指机体免疫系统对自身抗原不产生免疫应答,无免疫排斥的现象。
通常高等动物的免疫系统具有高度分辨“自己”与“非已”抗原物质的能力。在一般情况下,机体对“非已”抗原发生免疫排斥,而对自身抗原呈现自身耐受,其主要机制已如前述。
第一节 生理性自身免疫现象
1990年Ehrlich提出,在正常情况下,机体对自身抗原不产生免疫应答(即自身耐受性),只有当自身耐受遭到破环时才发生自身免疫。他曾认为机体一旦对自身抗原发生免疫应答,即将对机体造成严重的危害称之为自身中毒恐怖(horrorautotoxicus)。这个观点长期统治着免疫学领域,使自身免疫与自身免疫病两个概念等同起来。现代免疫学观点趋向于将这两种概念分开,对自身免疫的生理意义有许多崭新的认识。
现知自身免疫现象在正常人体内可起维持机体生理自稳的作用。正常人血清可以测得多种天然自身抗体,诸如抗肌动蛋白、肌凝蛋白、角蛋白、DNA、细胞色素C、胶原蛋白、髓鞘碱性蛋白、白蛋白、铁蛋白、IgG、细胞因子、激素等抗体,但这些抗体起着维持机体自稳的作用。体内产生的自身抗体有助于清除受损伤组织及其分解产物。
不同淋巴细胞克隆间的相互识别,在体内可构成独特型免疫网络,亦属于自身免疫现象,它在通常情况下起生理性免疫调节作用,使机体对外来抗原的应答有一定的自限性。
1975年Opelz等报道的自身混合淋巴细胞反应(autologous mixedlymphocytereaction,AMLR)是一种典型的自身免疫现象,已得到较深入的研究。AMLR系统中,自身的DR+细胞作为刺激细胞,CD+自身反应性T细胞(auto-ueactive Tcell)作为应答细胞被刺激而分裂增殖。早期增殖T细胞是TH,能分泌IL-2、B细胞生长因子、B细胞分化因子。晚期增殖细胞是TS或TC细胞。AMLR系统表明,机体在无外来抗原刺激下,自身反应性T细胞能识别自身抗原而产生辅助、抑制和杀伤效应。其中早期生成的TH细胞为诱导晚期TS,TC提供条件,而对被生物或理化因子作用而改变的自身细胞以及衰老蜕变的自身细胞有杀伤和清除作用。由此可见,AMLR对维持免疫自稳、自身耐受也有重要生理意义。
免疫应答过程各时相中自身MHC的限制作用更是说明机体在对外来抗原的识别和排斥时,均须以对自身抗原识别为基础。换句话说,在许多情况下,免疫细胞若不能识别自己,也就无法识别异已。
当自身免疫表现为质和量的异常,自身抗体和(或)自身致敏淋巴细胞攻击自身靶抗原细胞和组织,使其产生病理改变和功能障碍时,才形成自身免疫(autoimmune disease)。自身免疫与自身免疫病的关系可能有三种性况,①自身免疫引起疾病;②疾病引起自身免疫;③某些因素同时引前两者。
第二节 病理性自身免疫应答的诱因
一、自身抗原与佐剂的作用
(一)隐蔽抗原的释放
隐蔽抗原指体内某些与免疫系统在解剖位置上处于隔绝部位的抗原成分。按Burnet学说,由于这些抗原在胚胎期未曾与免疫系统发生过接触,故体内能与这些抗原起反应的免疫活性细胞未消失。在手术、外体或感染等性况下,隐蔽抗原释入血流或淋巴道与免疫系统接触,从而发生自身免疫。精子、眼晶状体、神经髓鞘磷脂碱性蛋白、某些器官、细胞(如甲状腺、胃壁细胞等)特异的微粒体抗原属此类型。实验证明,将眼晶状体或精子抗原注入动物自身体内,可诱发自身抗体。人体输精管结扎可形成抗自身精子的抗体以及眼球损伤后则可发生交感性眼炎。
(二)经改变的自身抗原
一系列生物、物理、化学因子均可使自身抗原成分改变以致引起自身免疫反应。微生物感染机体,破坏组织、细胞造成自身组织抗原改变,使之成为非已的物质而产生自身抗体。例如肺炎支原体感染可改变红细胞表面的Ⅰ血型抗原,产生抗红细胞的冷凝集素。又如在感染过程中,中性粒细胞吞噬细菌后释放出溶酶体酶,改变了自身IgG分子结构。这种变性的IgG可作为非已物质刺激机体产生抗IgG抗体,这种抗体主要是IgM,即类风湿因子。类风湿因子。类风湿因子在少部分正常人(老年人)中以及在很多慢性病患者体内均可发现,如梅毒、瘤型麻风、结核、乙型肝炎以及一些寄生虫感染。此外,溶酶体酶也可改变自身细胞表面抗原,使产生抗粒细胞抗体,引起粒细胞减少。
近年来人们注意到病毒感染与自身免疫的发生有密切关系,如风疹和乙型肝炎病毒感染后可发生血管炎和关节炎;巨细胞病毒和EB病毒感染可引起Coombs试验阳性的溶血性贫血;系统性红斑狼疮(SLE)患者血清中可测出抗麻疹病毒、EB病毒等抗体。
在病毒感染如乙型肝炎病人中还常发现抗核抗体、抗线粒体、抗平滑肌抗全等多种自身抗体。现已知病毒感染宿主时,在病毒进入细胞复制繁殖的过程中,病毒抗原能整合到宿主细胞表面,使宿主细胞膜抗原或组织相容性抗原、微丝肌动蛋白等成分发生改变。
化学药物引起自身抗原的改变也并不少见。如长期服用α-甲基多巴的患者中有10%~15%Coomba试验阳性,约有1%可发生自身溶血性贫血。这种病人在红细胞成熟过程中受药物影响改变了红细胞膜上的Rh系统的e抗原,产生抗红细胞抗体。又如长期服用肼苯达嗪、普鲁卡因酰胺、异烟肼等药物可引起红斑狼疮样综合症,常可测得抗核抗体。这些药物能与细胞内组蛋白或DNA结合,改变自身组织成分产生自身抗体。长期来观察到组织中损伤也可引起自身免疫反应,推测可能是由于组织抗原受到部分酶解,使天然分子中原处于隐匿部位的抗原决定簇暴露,被机体作为新抗株加以识别,这种看法有一定的实验根据,例如兔甲状腺球蛋白经胰酶消化处理后可使兔产生免疫反应,经白细胞蛋白酶消化的兔的甲状腺球蛋白也能引起兔的甲状腺炎。
(三)交叉抗原
某些微生物的抗原与自身组织成分有共同抗原性。因此在感染这些微生物后,机体所产生的抗体或致敏淋巴细胞对有关的自身组织也可产生免疫反应。如A组溶血性链球菌某些型别的胞壁抗原和胞浆膜抗原与人肾基底膜及心瓣膜成分相同,也与人心肌、骨骼肌、小动脉平滑肌相同,因此溶血性链球菌感染与风湿病和肾小球肾炎有密切关系(表16-1)。又如大肠杆菌O14与结肠粘膜有类似的抗原性,它与溃疡性结肠炎的发生有关。
最近研究注意到热休克蛋白(heat shock protein,HSP)或称应激蛋白(stress protein,SP)在诱导自身疫病发生中的作用。1926年Ritossa首先观察到正常果蝇暴露于高温后,其唾液腺染色体蓬松隆起。12年后,Tissieres等证实果蝇染色体遇热后的上述变化是由染色体内基因转录合成一种特定的蛋白质所致,遂提名为热休克蛋白。此后的研究发现,除温度外,各种应激因素,如感染、创伤、化学品等生物、理化因子、均可诱生这类蛋白合成增加,故又称应激蛋白。现知HSP存在于一切生物细胞,包括原核及真核细胞。按其分子量大小成分HSP90,HSP70,HDP60,小HSP四个家族,各含数个成员。HSP在进化上具有很大的保守性。因此其它物种来源的(各种病原体)HSP与人类的HSP(或其它抗原)有很大的相似性,或称分子模拟(molecularmimicry)。目前认为,感染诱导自身免疫病的重要原因之一是与HSP的参加有关。
表16-1 与人体组织起交叉反应的链球菌抗原
菌体抗原 |
细菌 |
人体组织抗原 |
组别 |
型别 |
细胞壁M蛋白 |
A |
1,5,19,29 |
心肌 |
胞壁蛋白-脂肪- |
A |
|
心肌 |
糖复合物 |
G |
除24型外 |
骨格肌 |
胞壁多糖体 |
A |
|
心瓣膜粘蛋白 |
胞膜糖蛋白 |
A |
|
肾小球基底膜 |
细胞膜 |
A |
12 |
组织相容性抗原 |
|
|
|
|
|
至今研究最多的为HSP60家族的作用,该家族成员包括分子量60KD上下的多个蛋白质。微生物的HSP65(HSP60家族)是优势蛋白,感染该微生物的人体中有40%T细胞应答是针对这一蛋白质的。该蛋白质有仅与人类HSP60相当,而且已经检出至于少与人类19个已知自身抗原的氨基酸序列有相似性,并与相应自身免疫的发生有关。仅举数例列表(16-2)。
表16-2 HSP60与人自身抗原相似性
HSP60区域 |
已知自身抗原 |
自身抗原区域 |
相似程度% |
有关疾病 |
1~17 |
髓过氧化物酶 |
594~610 |
60 |
肾小球肾炎 |
7~21 |
细胞色素P450 |
359~372 |
64 |
慢性活动性肝炎 |
65~75 |
甲状腺球蛋白 |
393~403 |
58 |
慢性甲状腺炎 |
86~100 |
肌凝蛋白重链 |
516~530 |
62 |
柯萨基心肌炎 |
101~123 |
细胞角蛋白 |
83~105 |
58 |
类风湿性关节炎 |
108~117 |
DNA-结合蛋白 |
73~82 |
58 |
SLE |
468~480 |
乙酰胆碱受体 |
133~145 |
63 |
重症肌无力 |
(四)非特异免疫细胞刺激剂
上述自身抗原仅激活相应抗原特异的淋巴细胞克隆。而非特异免疫细胞刺激剂,如佐剂、T、B细胞多克隆激活剂和近年来发现的超抗原,通过多克隆激活静止的自身反应性T、B细胞,促进自身免疫应答甚至自身免疫病的形成。福氏完全佐剂联合自身组织匀浆或已知纯化自身抗原被广泛用于诱发实验性自身免疫病的方法并获得满意结果。
二、机体因素
内因包括机体内的各种重要条件因素,相同外因作用下,内因条件不同,后果也不同。
(一)遗传因素
临床上早就发现自身免疫性疾病的发生有家族史倾向。在动物实验研究中陆续发现了几种自身免疫病的品(NZB、NAB/NAWF1小鼠OS来亨鸡等)进一步提示遗传与自身免疫病的关系。70年代以来,对人白细胞抗原(HLA)的研究有了很大的进展。发现HLA与某些疾病,特别是与某些自身免疫性疾病的发生密切相关。如患有强直性脊椎炎的病人中90%以上带有HLA-B27抗原。凡带有HLA-B8、DW3、DRW3单体型的人发生多种自身免疫病的危险性比带有其他型的人高得多。Addison病与B8、DR3有关,胰岛素依赖型糖尿病(又称型或青年型糖尿病)与B8、DR3、DDR4、DR3/4有关,类风湿关节炎与DR4有关。慢性甲状腺炎(Hashimoto病HLA-DR5有关。新近,人们对MHC与疾病关联的研究深入到了分子水平。糖尿病低抗者DQβ链第57位是天门冬酸,而易感者常出现丝氨酸、缬氨酸、丙氨酸。在NOD(nonobese diabetic)转基因小鼠中证实了上述结果。
利用小鼠胰岛素依赖型糖尿病(IDDM)模型研究发现,疾病发生受多基因控制。基因之一是与H-2K端相连锁。第二个糖尿病基因座在第9对染色体位于Thy-1与MDD1基因标志之间。通过返交试验发现易感动物至少有3个隐性致糖尿病基因。
遗传易感倾向并非绝对,疾病的外显率也受环境因素影响。
(二)机体免疫系统功能失常
统计数字表明体液或细胞免疫缺陷者自身免疫病的发生率高于正常随机人群。先天性或后天获得性低丙球蛋白血症者自身免疫病的发生率可高达14%,而正常人群(指无免疫缺陷)中的发生率为0,001~0.01%。细胞免疫缺陷常同时伴有肿瘤与自身免疫的高发现象。
胸腺功能不全、慢性病毒感染、增生性变化等与自身免疫的发生有关。SLE患者血清中胸腺激素含量下降,胸腺萎缩,试用胸腺素治疗曾观察到症状缓解。摘除胸腺对重症肌无力者有显著疗效。
(三)年龄、性别及内分泌的影响
临床观察老年人中自身抗体检出率增高,在60~70岁以上的老年人中有50%以上可以检出自身抗体。临床观察也发现自身免疫病多好发于妇女,据统计SLE患者,女性比男性多9倍以上。动物实验证实了性激素在自身免疫病发展中的作用。将雄性NZB小鼠进行阉割,可加速病变、病情加剧、寿命缩短。而雌性病鼠接受雄激素治疗则可延长寿命。
第三节 病理性自身免疫发生机制
目前认为上述诱因导致自身免疫的形成是通过多种机制而实现的。不同情况下由不同机制起主要作用,有时也可能由几种机制同时或先后起作用。
一、禁忌株突变
通过体细胞突变可能产生被Burnet称为禁忌的细胞株。使在正常情况下已遭克隆清除的自身反应性T.B细胞再现。
二、T、B细胞活化信号的出现
正常人体内存在一些自身反应性T.B细胞克隆,因缺乏足够的激活信号而处于无应答状态(失活或静止状态)。一旦获得足够的激活信号,便可产生正相应答。
现有实验证明,在某些自身免疫病中受累器官细胞表面,或是出现了原先不表达的DR/I-A抗原,或是DR/IA的抗原较原先表达增多。病毒感染导致自身免疫病的可能机制是通过机体释放的淋巴因子,如IFN-r和IL-1等。使得原先不表达MHC-Ⅱ类抗原的组织细胞出现DR/I-A抗原。同种免疫可以诱导出自身免疫,可能亦与此机制有关。例如造成GVH病动物体内角质细胞表面表达I-A抗原,I-A阳性T细胞增加。这些质和量改变的MHCⅡ类抗原可激发自身免疫反应。实验证明,注射抗H-21区抗原的抗体可预防或减弱小鼠实验性重症肌无力、脑脊髓和Ⅱ型胶原性关节炎等。
三、B细胞被多克隆激活
在T细胞处于耐受的情况下,用B细胞多克隆激活剂直接刺激B细胞,可导致自身抗体的产生。例如,给小鼠注射LPS与自身抗原并用,则可导致特异性自身抗体生成。甲状腺球蛋白与LPS合用可诱发抗甲状腺球蛋白特异抗体。
四、TH细胞旁路激活
此学说的基本观点是机体对某些自身抗原的耐受性仅是由于T细胞处于耐受状态,使B细胞缺少辅助信号而不能有效地活化。一旦通过旁路机制获取T细胞辅助信号,B细胞即可活化,产生自身抗体(图16-1,2)
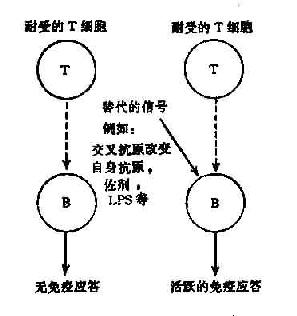
图16-1 T细胞旁路活化机制
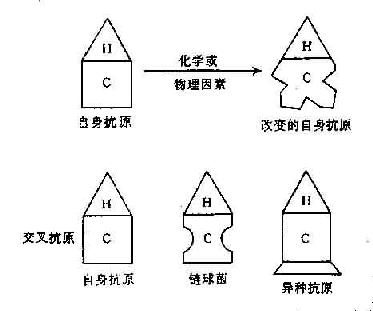
图16-2 破坏T细胞对自身抗原的耐受性
佐剂、病毒、细胞产物、以及某些药物具有非特异地激活T、B细胞的作用(多克隆激活),它们诱发自身免疫的机制可能就是通过替代因子的旁路作用,绕过耐受的T细胞,使B细胞活化。
T细胞旁路机制不但可诱导自身抗体生成,也能引起细胞介导的自身免疫性损伤,如实验性变态反应性脑脊髓炎。甲状腺炎等。
五、自身反应克隆脱抑制
TS细胞能抑制自身反应细胞的激活,TS细胞数量或功能降低,TH和(或)TCS细胞数量增多或活跃,使自身反应细胞发生脱抑制而功能亢进,都可导致自身免疫的发生。
早期系统的实验资料曾对SLE样综合征自发品系NZB及NAB/NZW小鼠自出生后逐月进行免疫功能检测。从时间顺看,这种动物体内最早出现的是T细胞调节功能紊乱,约在出生后1个月,动物血清中胸腺激素活性下降,TS细胞功能减退。T细胞对诱导免疫耐受呈抵抗,然后再出现B细胞功能亢进。接近3月龄时,血清中才出现抗核抗体、抗淋巴细胞表面抗体及其他抗血细胞抗体等多种自身抗体。
受动物实验启示,临床免疫工作者测定各种自身免疫病患者外周血T细胞亚群变化,也发现类似变化。应用抗T细胞分化抗原单克隆抗体测定时,发现SLE和类风湿关节炎患者在病性活动期,TS细胞比例下降,用Con A在体外刺激病人淋巴细胞不诱发非特异的TS细胞。
六、独特型网络激活
独特型-抗独特型的相互作用极为复杂。在不同的条件下,对自身免疫产生不同的调节作用,有时抑制(自身耐受),有时增强。外来或体自身抗原均可通过此网络活化而造成自身免疫。根据独特型网络的基本理论,有以下几种可能性(图16-3):

图16-3 独特型相互作用形成自身免疫
1.针对抗原(细菌、病毒、激素等)所产生的抗体,具有与自身细胞膜抗原相似或相同的决定簇,故其所激发的抗独特型抗体能与自身细胞表面抗原结事(图16-3a)。已有实验证据揭示这种可能性的存在,如小鼠注射胰岛素后所产生的抗体能进一步诱发生成抗独特型抗体。这种抗体不仅能与激发抗体结合,也能与胰岛素受体结合,造成胰岛素抵抗型糖尿病。
2.抗微生物抗体本身可能就是一种抗自身抗体的抗独特型抗体,它能激活具有独特表位的自身的反应性B细胞(图16-3b)。
第四节 自身免疫性疾病
一、自身免疫性疾病的基本特征
自身免疫性疾病往往同时具有以下特点:①患者血液中可测行高效价自身抗体和(或)自身组织成分起反应的致敏淋巴细胞。②自身抗体和(或)自身致敏淋巴细胞作用于靶抗原所在组织、细胞,造成相应组织器官的病理性损伤和功能障碍。换句话说,患者组织器官损伤的范围取决于自身抗体或致敏淋巴细胞所针对的自身抗原分布格局。③在动物实验中可复制出相似的病理模型,并能通过患者的血清或淋巴细胞使疾病被动转移。④病情转归与自身免疫反应强度密切相关。⑤除一些病因明了的继发性自免疫性疾病可随原发疾病的治愈而消退外,多数原因不明的自身免疫常呈反复发作和慢性迁延。⑥疾病的发生有一定的遗传倾向。
二、自身免疫性疾病的分类
自从60年代初公认存着自身免疫性疾病以来,现已确认属于这种类型的疾病有30余种,其分类方法主要有二。其一是按疾病累及的系统区分(表16-3);其二是按器官特异性分为两类(表16-4)。
表16-3各系统自身免疫病
不同系统疾病 |
自身免疫病举例 |
结缔组织疾病 |
类风湿关节炎、系统性红斑狼疮、皮肌炎、硬皮病 |
神经肌肉疾病 |
多发性硬化症、重症肌无力、脱髓鞘疾病 |
内分泌性疾病 |
原发性肾上腺皮质萎缩、慢性甲状炎、青少年型糖尿病 |
消化系统疾病 |
慢性非特异性溃疡性结肠炎、慢性活动性肝炎、恶习性贫血与萎缩性胃炎 |
泌尿系统疾病 |
自身免疫性肾小球肾炎、肺肾出血性综合症 |
血液系统疾病 |
自身免疫性溶血性贫血、特发性血小板减少性紫癜、特发性白细胞减少症 |
图16-4 两类常见的自身免疫病及其相应的自身抗原
自身免疫病 |
自身抗原 |
器官特异性自身免疫病: |
|
慢性甲状腺炎 |
甲状腺球蛋白、微粒体、细胞膜表面抗原、第二胶质抗原(CA2) |
毒性弥漫性甲状腺肿(Graves病) |
甲状腺细胞表面TSH受体 |
原发性肾上腺皮质萎缩( Addison病) |
肾上腺细胞 |
恶性贫血 |
胃壁细胞、内因子 |
慢性溃疡性结肠炎 |
结肠上皮细胞 |
男性自了性不育症 |
精子 |
青少年型胰岛素依赖性糖尿病 |
胰岛细胞 |
伴共济失调-毛细血管扩张的胰岛素低抗型糖尿病 |
胰岛素受体 |
重症肌无力 |
乙酰胆碱受体 |
自身免疫性溶血性贫血 |
红细胞 |
特发性血小板减少性紫癜 |
血小板 |
干燥综合征(Sjogrens syndrome) |
唾液腺管、细胞核、甲状腺球蛋白 |
非器官特异性自身免疫病: |
|
类风湿关节炎 |
变性IgG |
系统红斑狼疮 |
核成分(DNA、DNA-核蛋白、RNA、Sm抗原)、红细胞、血小板、细胞浆成分(线粒体、微粒体) |
三、自身免疫病组织损伤机制
已有大量的资料证明,自身免疫病理损伤是由自身免疫应答的产物包括自身抗体和(或)自身致敏淋巴细胞引起的,后者造成病理损伤的机制与各型超敏反应相同(表16-5),以Ⅱ型至v型多见。Ⅱ型超敏反应中,自身抗体与细胞膜或基底膜自身抗原结合,在膜表面形成免疫复合物。后者通过结合并激活补体链锁反应,在膜表面出现补体活化反应产物C56789,对膜产生破坏性攻击,造成靶细胞裂解或基底膜损伤。此外覆盖免疫复合物的靶细胞被吞噬细胞加速吞噬(免疫调理)以及K细胞进行非天噬性杀伤。Ⅲ型超敏反应中,自身抗体与自身抗原在血循环中相遇,形成免疫复合物,在一定的物件下,沉积于相应部位的组织间隙。局部免疫复合物结合并激活补体,补体活化反应产物导致局部发生炎症反应。N型超敏反应中,自身致敏淋巴细胞攻击局部靶组织,造成局部炎症。V型超敏反应则由自身抗体结合并刺激靶细胞,使其功能亢进。
表16-5 自身疫应答产物造成病理损伤的机制(举例)
自身免疫病 |
自身免疫应答产物 |
超敏反应类型 |
病损特征 |
(Goodpasture综合症) |
抗肾小球,肺泡基底膜N胶原 |
Ⅱ |
肾炎,伴蛋白尿、肺出血 |
自身免疫性溶血性贫血 |
抗红细胞膜蛋白抗体 |
Ⅱ |
溶血 |
自身免疫性血小板减少性紫癜 |
抗血小板膜蛋白、如gpⅡb/Ⅲa抗体 |
Ⅱ |
血小板破环,减少 |
重症肌无力 |
抗神经肌肉接头处乙酰胆碱受体的抗体和自身致敏淋巴细胞 |
Ⅱ.Ⅳ(?) |
乙酰胆碱受体破坏,神经冲动传递低下,肌无力 |
类风湿性关节炎 |
抗IgG抗体(类风湿因子)抗HSP致敏淋巴细胞(?) |
Ⅲ.N(?) |
关节腔炎症 |
系统性红斑狼疮 |
抗DNA、核蛋白、各种血细胞膜抗原等抗体 |
Ⅱ. Ⅲ |
血细胞减数,多部位(肾、关节、血管)炎症 |
实验性变态反应性脑脊髓炎 |
抗髓碱性蛋白之自身致敏T细胞 |
Ⅳ |
脑脊髓炎症 |
实验性变态反应性神经炎 |
抗外周鞘碱性蛋白之自身致敏T细胞 |
Ⅳ |
神经炎 |
某些自身免疫性甲状腺炎 |
抗甲状腺滤泡上皮细胞之致敏T细胞 |
Ⅳ Ⅳ |
甲状腺炎 |
甲状腺功能亢进 |
抗TSH受体抗体 |
Ⅴ |
甲状腺细胞分泌甲状腺素增加 |
第五节 自身免疫病治疗原则
自身免疫病的治疗尚缺乏理想的方法。通常针对疾病的病理变化和组织损伤所致的后果进行治疗,也可通过调节免疫应答的各个环节阻断疾病进程来达到治疗的目的。
(一)抗炎药物
大剂量皮质激素的应用可有效地抑制一些重症自身免疫病所致的炎症反应。其它抗炎药物如水杨酸制剂、各种合成的前列腺素抑制剂等也广泛采用。淋巴因子和补体的拮抗剂亦有利于抑制炎症反应。
(二)免疫抑制剂
环孢素A(cycrosporinA)是目前一种广为推荐的免疫抑制剂,它是一种不溶性的真菌代谢产物,能有效地抑制T细胞介导的细胞免疫反应。对T细胞的作用主要是抑制某些基因特别是IL-2基因的转录,从而阻断IL-2的合成和分泌,使T细胞的扩增和分化受阻。它还能抑制c-myc和IFN-γ基因的转录,后者对于T细胞的活化和扩增也有影响。环孢素A是一种兼有抗有丝分裂和抗炎效应的免疫抑制剂。已证实它对眼色素层炎、早期Ⅰ型糖尿病、肾病综合症、牛皮癣等有较好的疗效,对特发性血小板减少性紫癜、系统性红斑狼疮、多发性肌炎、Crohn病、原发性胆汁性肝硬化、重症肌无力症、类风湿性能关节炎均有一定的治疗效果。FK-506是继环孢素A后发现的另一种真菌代谢物,其结构与环孢素A不同,但它的作用与环孢素A极为相似。FK-506应用剂量较低,故其副作用较小。其它的抗有丝分裂的非特异性免疫抑制剂如硫唑嘌呤、环磷酰胺、环磷酰胺、氨甲喋呤常与皮质激素联合应用作为常规免疫抑制剂治疗一些自身免疫病。
(三)免疫调节
它是根据调节免疫应答规律以达到阻断自身免疫过程提出的一种治疗设想。包括下列的一些措施。
1.清除或使某些免疫活性细胞失活实验研究发现,体内应用抗MHCⅡ分子与抗CD4单克隆抗体,可减轻系统性红斑狼疮和类风湿性关节炎的发展。
2.独特型的抑制
(1)抗体的调控抗独特型抗体在调节外来抗原诱发的抗体生成起重要作用,它可能对自身抗体的生成起抑制作用。
(2)T细胞疫苗 给动物注射髓鞘碱性蛋白特异的碱活T细胞克隆(亚致病剂量),能有效地预防实验性变态反应性脑脊髓炎的发生。这可能是通过诱导生成针对效应T细胞受体独特型的抑制性T细胞所致。
(3)抗原封阻或清除相应的自身反应性淋巴细胞已有实验证明,赃与自身抗原类似的多肽片段,同自身抗原竞争性地结合到抗原呈递细胞的MHC分子上,阻断自身抗原诱发的T细胞应答,达到治疗自身免疫性变态反应性脑脊髓炎的效果。也有使用导向技术,将各处毒素或放射性物质偶联到自身抗原上,以此偶联物选择性地杀灭特异的自身反应性淋巴细胞。此外,有人致力于研究出针对自身抗原特异的抑制因子提供临床使用。
(四)血浆置换
此疗法的目的在于降低自身免疫病人血浆中的免疫复合物的含量,减轻免疫复合物在组织中沉积。对于是治疗有生命威胁的免疫复合物所致的血管炎、系统红斑狼疮、肺肾出血性综合征等有一定的治疗效果。若与抗有丝分裂的药物联合应用不着,疗效更佳。
(五)对症治疗
通常在治疗某些器官特异性的自身免疫病时,祗需调整器官损伤所造成的代谢障碍,即可达到控制病情的效果。如自身免疫性甲状腺炎的粘液性水肿患者可采用甲状腺替代疗法,青年型糖尿病患者用胰岛素控制血糖,恶性贫血患者用维生素B12,甲状腺功能亢进者用抗甲状腺药物等。
